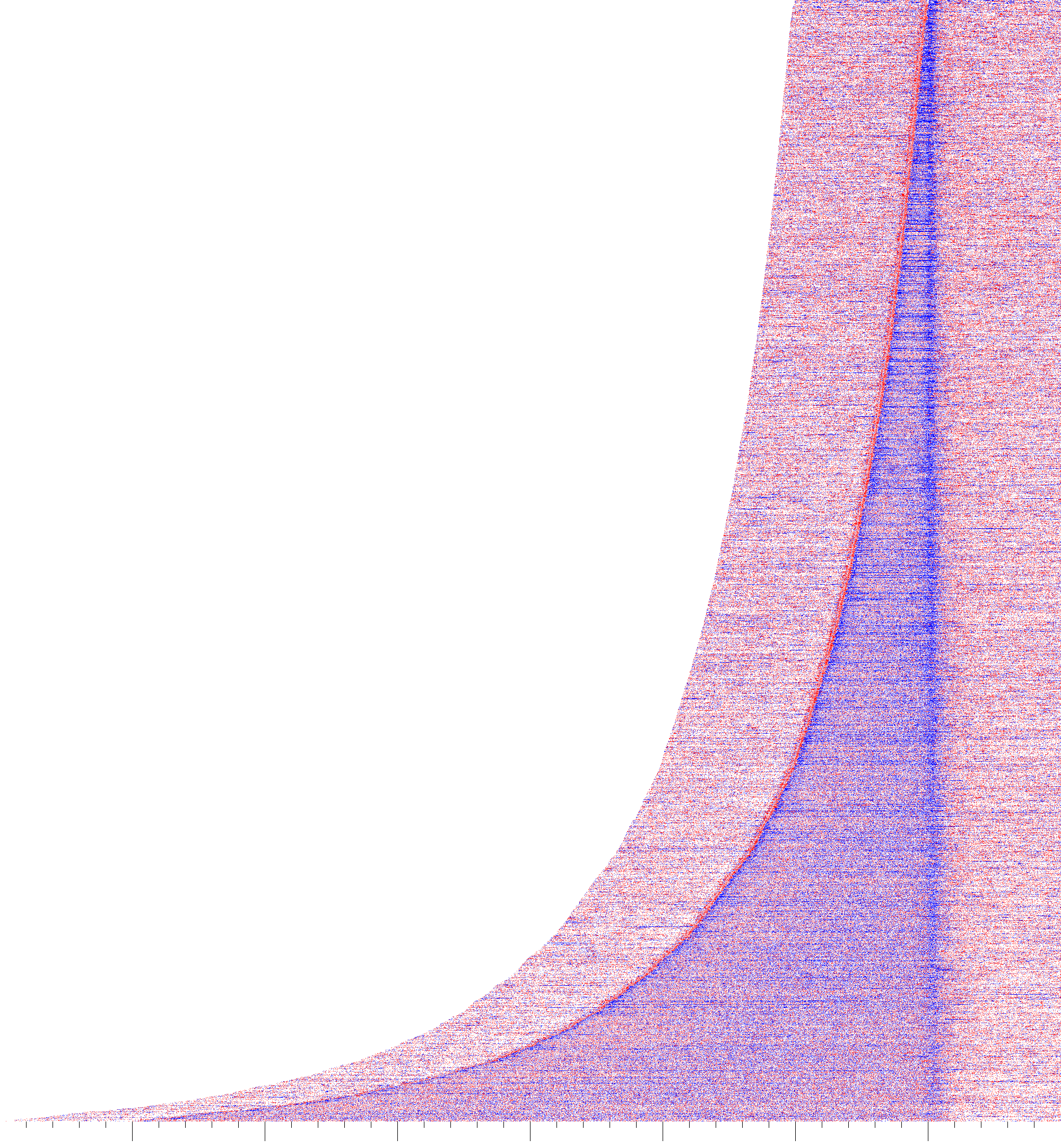
TES Fold Change Heatmap
The TES_heatmap tool generates a heatmap plotting the position and quantity of 3’ end reads sorted by gene length centered on the TES.
Note
This tool requires bedtools to be installed.
Usage
Usage:
PolTools TES_fold_change_heatmap [-h] --numerator seq_file spike_in
--denominator seq_file spike_in
[-w width] [-e height]
[-d downstream_distance]
[-u upstream_distance] [-b bp_width]
[-m max_log2_fc]
[--minor_ticks minor_ticks]
[--major_ticks major_ticks]
[-t [threads]]
truQuant_output_file output_prefix
Required Arguments |
Description |
|---|---|
truQuant Output File |
File ending in -truQuant_output.txt generated from truQuant |
–numerator seq_file spike_in |
Sequencing file and its accompanying normalization factor to be used as the numerator of the heatmap. Additional files can be provided with multiple –numerator arguments |
–denominator seq_file spike_in |
Sequencing file and its accompanying normalization factor to be used as the denominator of the heatmap. Additional files can be provided with multiple –denominator arguments |
Output Prefix |
Output filename will begin with the output prefix and also contain the run parameters and ends in gene_body_heatmap.tiff. |
Optional Arguments |
Description |
|---|---|
-u, –upstream_distance |
Distance upstream of the max TSS from truQuant to show on the heatmap. Default is 50,000 bp. |
-d, –distance_past_tes |
Distance past the TES to show on the heatmap. Default is 50,000 bp. |
-b, –bp_width |
Total distance shown on the heatmap. Default is 400,000 bp. |
-w, –width |
Width of the heatmap in pixels. Default is 2,000 px. |
-e, –height |
Height of the heatmap in pixels. Default is 2,000 px. |
-m, –max_black |
Maximum value to consider as black. Default is the max value found. Decreasing this number will make the image darker. |
–minor_ticks |
Distance in bp between minor tick marks. Default is 10,000 bp. |
–major_ticks |
Distance in bp between major tick marks. Default is 50,000 bp. |
Behavior
TES_heatmap will create a file starting with the output prefix which contains the run parameters and ends in TES_heatmap.tiff.
First, regions to be quantified are designed from a distance upstream of the max TSS
to a distance downstream of the TES (default is 50 kb away from each position) for each gene in the truQuant file. Then,
the tsrFinder run associated with
truQuant is used to find regions to blacklist
(TSRs with at least 30% of the number of 5’ ends found in the pause region of the gene). These regions are blacklisted
in the sequencing files and the 3’ ends are quantified in each location of the previously defined regions. The 3’ end
reads are normalized according to the correction factors. The two numerator datasets are added together and divided by
the sum of the denominator datasets. The resulting division matrix is subjected to a log2 fold change transformation, to
generate the heatmap.
For example:
$ head seq_file-truQuant_output.txt
Gene Chromosome Pause Region Left Pause Region Right Strand Total 5' Reads MaxTSS MaxTSS 5' Reads Weighted Pause Region Center STDEV of TSSs Gene Body Left Gene Body Right Gene Body Distance seq_file.bed Pause Region seq_file.bed Gene Body
NOC2L chr1 959177 959327 - 194 959255 46 959250 13.306459171023036 944203 959177 14974 194 18
KLHL17 chr1 960552 960702 + 234 960632 27 960626 25.417791063821863 960702 965719 5017 234 17
PLEKHN1 chr1 966439 966589 + 25 966521 8 966513 19.47408534437497 966589 975865 9276 25 11
HES4 chr1 1000013 1000163 - 239 1000096 87 1000086 27.14758979723915 998962 1000013 1051 239 68
ISG15 chr1 1000204 1000354 + 160 1000295 12 1000278 36.24344768368484 1000354 1014540 14186 160 111
AGRN chr1 1020042 1020192 + 112 1020119 35 1020116 25.189637892253575 1020192 1056118 35926 112 76
RNF223 chr1 1074208 1074358 - 32 1074306 10 1074284 32.567238138964136 1070967 1074208 3241 32 8
C1orf159 chr1 1116028 1116178 - 51 1116106 9 1116103 19.81136532595448 1081818 1116028 34210 51 11
SDF4 chr1 1231907 1232057 - 1105 1231971 321 1231978 23.701136922154493 1216908 1231907 14999 1097 177
$ PolTools TES_fold_change_heatmap seq_file-truQuant_output.txt --numerator flavo.bed 1 --denominator dmso.bed 1 treatment -m 2